The SARS-CoV-2 main protease is a target for the development of novel therapeutics for COVID-19. Based on crystal structure data from the homologous SARS-CoV-1 protease, the binding site would be expected to adopt two possible conformations – a close-apo-like structure with the His-Cys catalytic dyad intact, and an open structure with the His-Cys dyad reorganised in such a way to facilitate ligand binding between these residues.
In this manuscript, Mohamed Ali Al-Badri, from our group, and Khaled Abdel-Masoud, who is a PhD student in Prof. Jonathan Essex’s group at the University of Southampton, have conducted advanced enhanced sampling molecular dynamics simulations to explore the equilibrium between the open and closed binding site forms. In doing so, we have foundthat the lower bound on the free energy required to open the binding pocket is 4 kJ.mol. This free energy penalty may readily be recovered through favourable protein ligand interactions.
We therefore recommend that efforts to identify inhibitors to the SARS-CoV-2 main protease should consider both open and closed structural forms. More generally, this study demonstrates the power of molecular simulation in guiding drug discovery efforts.
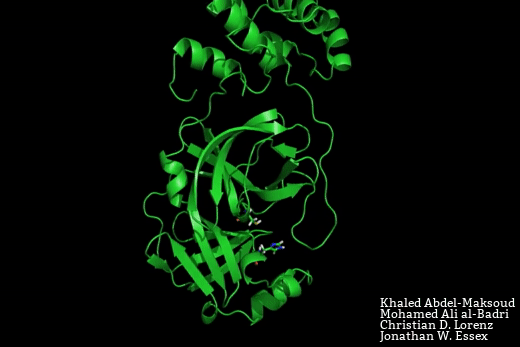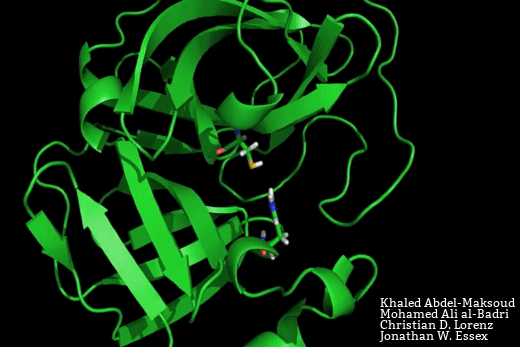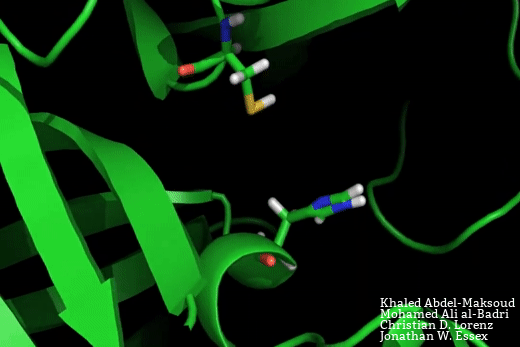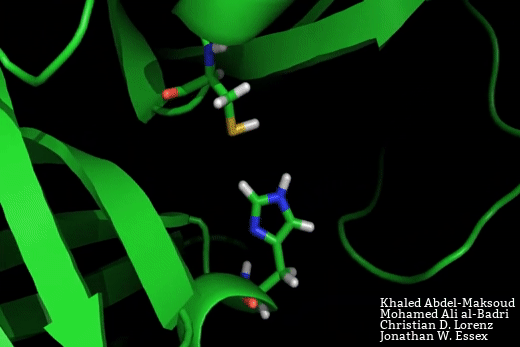
Full reference: “Allosteric Regulation of SARS-CoV-2 Protease: Towards Informed Structure-Based Drug Discovery,” Khaled Abdel-Maksoud, Mohamed Ali al-Badri, Christian Lorenz & Jonathan W. Essex (2020) chemRxiv.