In this work, we demonstrate to atomic precision the structural origins of protein corona formation due to adsorption, driven only by the choice of functional groups on a graphitic material. Using graphene-oxide (GO) and double-clickable azide- and alkyne-double functionalised graphene oxide (C2GO) sheets, we study the adsorption of apolipoprotein c-3 (apo-c3).
Our results detail the denaturing of GO-adsorbed apo-c3 on the hundreds of nanoseconds scale, following which its tertiary structure is stabilised by forming protein-protein binding motifs in the form of β-bridges. Whereas C2GO-adsorbed apo-c3 largely retains its tertiary structure and importantly retains the secondary structure of its C-terminus, conserving its primary function in lipid binding and potentially enabling cellular uptake of the protein- nanomaterial complex.
We present an investigation using molecular dynamics simulations of protein adsorption on graphitic materials, aided by a comprehensive analysis pipeline involving dimensionality reduction and machine learning clustering, binding free energy calculations and various protein structure analyses. To the best of our knowledge, this is the first application of such a pipeline to the study of protein adsorption on nanomaterials. This method can therefore provide valuable insight to the study of protein corona formation on any nanomaterial of interest, extracting dynamic features that underpin protein corona formation.
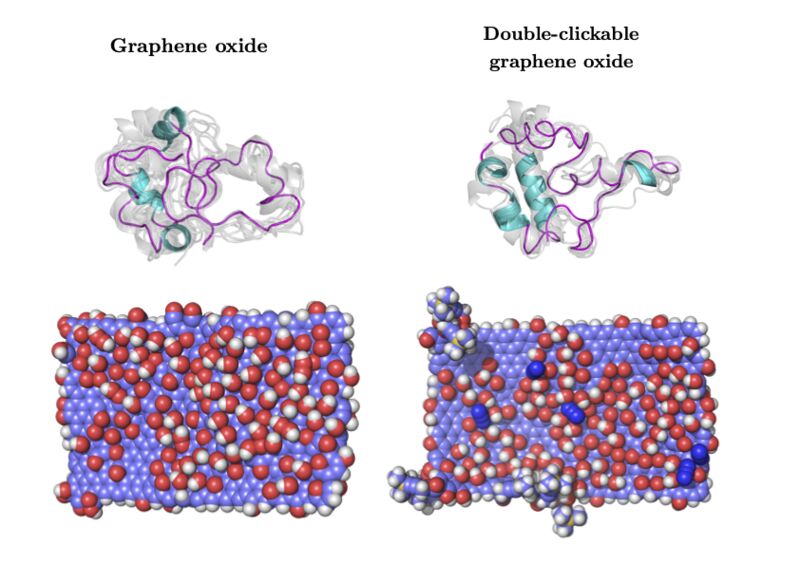
Full reference: Nanomaterial functionalization modulates hard protein corona formation: Atomistic simulations applied to graphitic materials. Mohamed Ali al-Badri, Paul Smith, Khuloud T. al-Jamal & Christian D. Lorenz, Advanced Materials Interfaces (2022) 9 (1), 2101236.